Life SciencesEconomic Overview & Market Update
Oct 29, 2025
Heading into 2025, the life sciences industry had an optimistic financial outlook but braced for market and business volatility due to the new U.S. administration, pending global regulatory changes, and geopolitical uncertainties. While the top focus of the pharmaceutical industry in the U.S. was the Inflation Reduction Act, additional regulation of software as a medical device, the overturn of the Chevron doctrine, and the implementation of tariffs have industry-wide implications.
Deal maker confidence has been influenced by factors such as ongoing trade wars, tariffs, the Most Favored Nation prescription drug pricing executive order, and the impacts of budget cuts made to the Food and Drug Administration (FDA) by the Department of Government Efficiency. The impacts of the confirmations of Dr. Martin Adel Makary as the new Commissioner of the Food and Drug Administration and Robert F. Kennedy Junior as Health Secretary are developing.
The life sciences industry saw a modest year-over-year increase in mergers and acquisitions (M&A) activity in 2024, which economic forecasters expected to see continue and accelerate in 2025. The industry also saw a modest increase in overall capital markets activity in 2024, including an estimated 55% increase in initial public offerings, which was off a lower base to start, but was expected to gain momentum in the coming year as well.1
Deal activity in the pharmaceutical and life sciences sectors for the first half of 2025 remained consistent. Growth was led by targeted, strategic acquisitions in the $1 to $5 billion range. Refer to reference page for detailed list of the top ten deals in 2025.
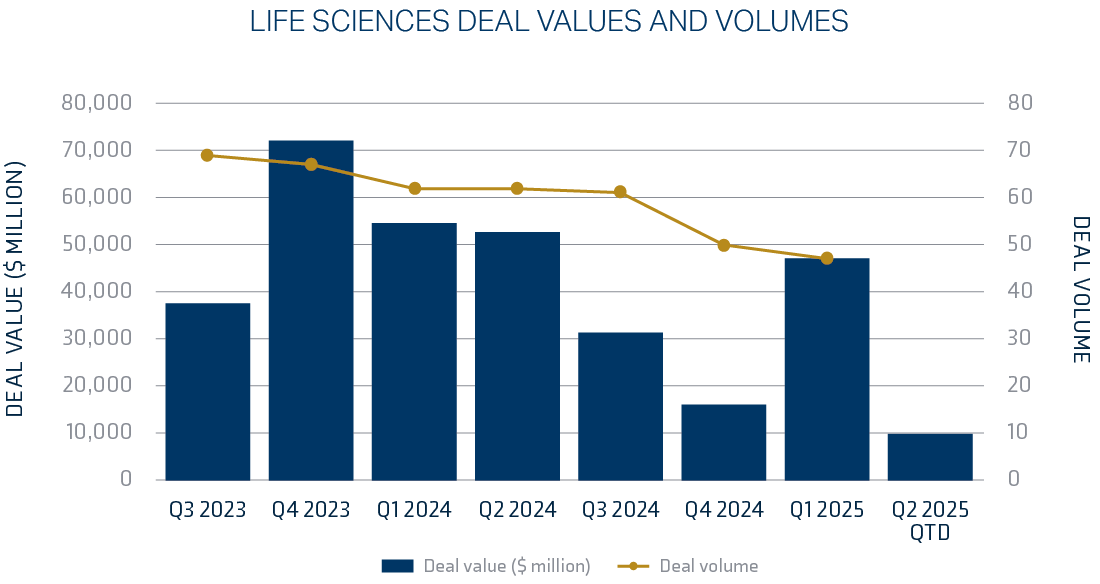
Overall, near-term prospects for M&A in the life sciences sector are expected to remain robust, given the level of cash available in the equity market, the number of upcoming drug approvals, and advancements in the adoption of advanced technologies, such as artificial intelligence.
The medical device market in 2025 is undergoing a significant transformation due to technological advancements, regulatory updates, and evolving patient needs, with projections indicating growth to $1.3 trillion by 2029.3 These changes are reshaping the industry and creating opportunities for innovation and improved patient care.
Technological Advancements: Artificial Intelligence and Machine Learning
Artificial intelligence (AI) and machine learning are transforming medical devices. These technologies equip devices to process vast datasets and deliver insights that improve diagnostics, treatment planning, and patient monitoring. AI-driven imaging systems, predictive analytics for disease management, and personalized treatment protocols are becoming more widespread.
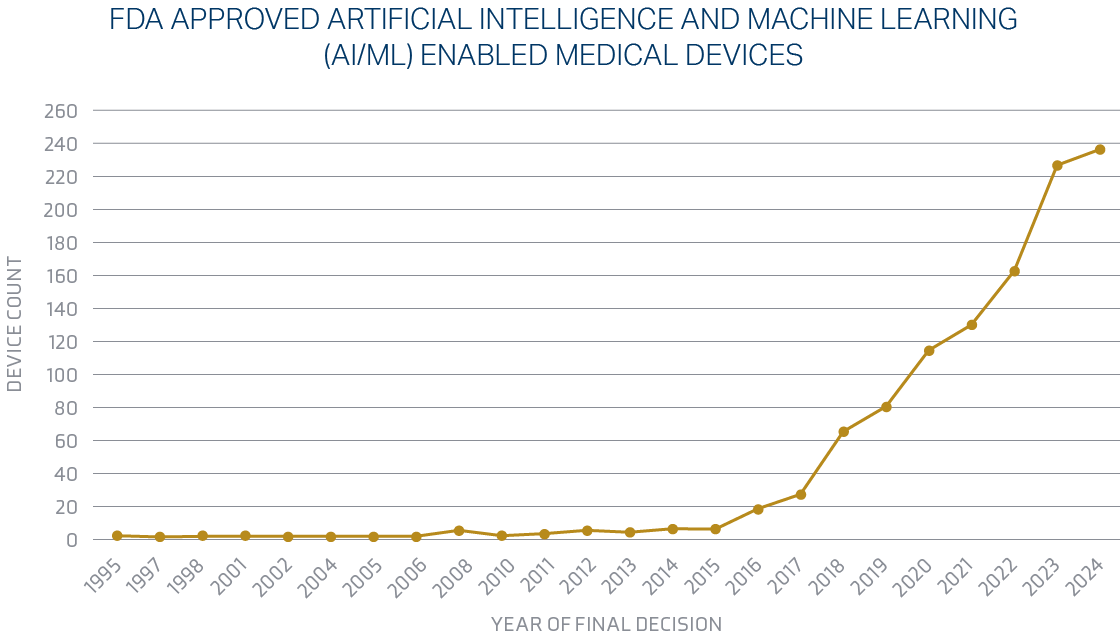
The Food and Drug Administration is making significant changes in 2025 regarding artificial intelligence for the medical device industry. The FDA published a draft guidance document on Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations in January 2025.4 A finalized guidance document on Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions was also released.5 This plan aims to facilitate iterative improvements to AI algorithms while maintaining regulatory compliance. Heading into 2025, the Food and Drug Administration passed a significant milestone, authorizing more than 1,000 devices enabled by artificial intelligence and machine learning. The FDA is currently clearing approximately 20 AI algorithms monthly, with an expectation that this number will increase.
Supply Chain
The medical device market is experiencing significant growth. This growth underscores the importance of a robust and efficient supply chain to meet increasing demand. In 2025, the Food and Drug Administration is focusing on strengthening the medical device supply chain, addressing potential shortages, and improving transparency. Key areas include enhanced reporting requirements for permanent discontinuance or interruptions. These changes aim to ensure a more resilient and efficient supply chain for medical devices, thereby improving public health outcomes. The FDA issued a final guidance document: Notifying the FDA of a Permanent Discontinuance of Interruption in Manufacturing of a Device under Section 506J of the FD&C Act.7
This guidance also provides a list of devices, by FDA product code,8 for which a manufacturer of such devices is required to notify the FDA in accordance with section 506J. The guidance also clarifies that the FDA may receive additional voluntary notifications regarding supply chain issues at any time, unrelated to the declaration or potential declaration of a public health emergency.
Early in 2025, there was a clear emphasis on supply chain and early identification of shortages. This focus has since shifted amid new trade policies and tariff implementation, which are expected to influence the medical device industry — a sector where approximately 69% of devices marketed in the U.S. are manufactured solely outside the country.9
Pharmaceutical funding trends in 2025 show a significant downward shift after a strong January start, with a sustained decline in private investment through July and muted public funding activity in the second quarter. While investment faces headwinds from policy uncertainty and capital constraints, strategic M&A is expected in the mid-range ($5 billion to $15 billion), and a surge in AI adoption, along with a focus on high-demand therapeutic areas such as weight management and oncology, continues to drive funding for innovation and breakthrough discoveries. By the end of 2025, AI is projected to generate between $350 billion and $410 billion annually for the pharmaceutical industry.
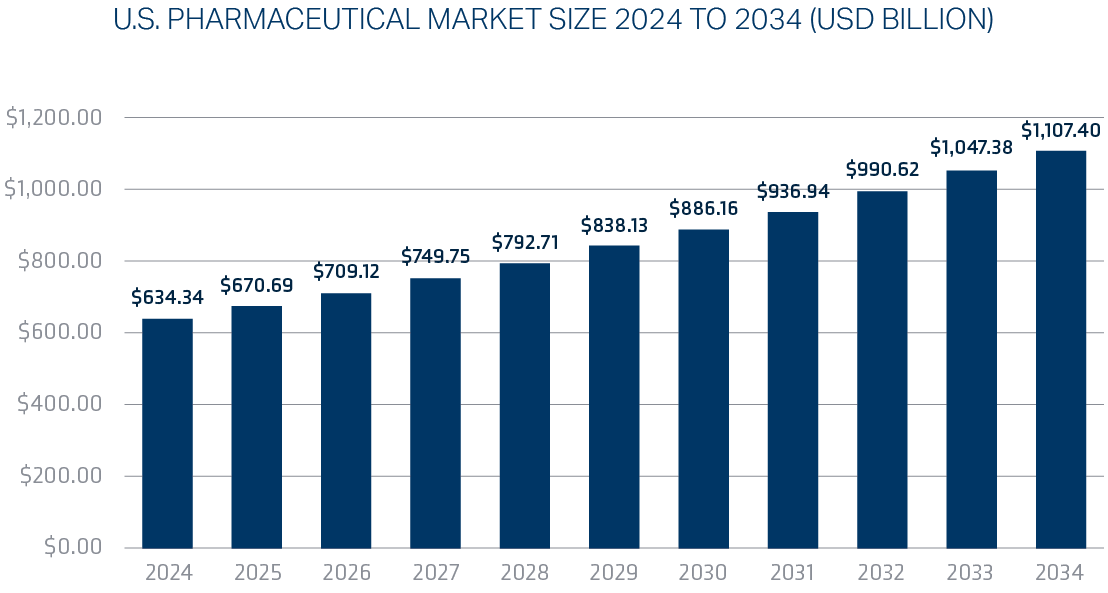
The U.S. pharmaceutical market size is calculated at $634.34 billion in 2024 and is predicted to attain around $1.107 trillion by 2034, expanding at a CAGR of 5.73% from 2025 to 2034.
Pre-Clinical Regulatory Changes
In the midst of widespread layoffs within the Food and Drug Administration in the first half of the year,11 the current administration is seeking ways to accelerate the drug approval process. In April, Dr. Martin Makary, head of the FDA, announced a plan to phase out animal testing requirements for monoclonal antibodies and other drugs. This is a groundbreaking advancement for the pharmaceutical industry in an effort to accelerate the drug approval process. Animal testing has long been a financial burden for drug development companies, in addition to being a time-consuming process that creates delays in getting new drugs to market.
The FDA’s animal testing requirements will be reduced, refined, or, potentially, replaced using AI-based computational models and organoid testing in laboratory settings. The implementation of the change took effect immediately for investigational new drug applications, with the FDA encouraging the adoption of the new suggested alternatives.
In addition, to aid in the determination of a drug’s efficacy, the agency will incorporate real-world safety data from other countries with comparable regulatory standards that have already studied the drug in humans.12
While the financial and scientific impacts of this monumental change are yet to be seen, the Food and Drug Administration hopes that easing the animal testing burden on pharma companies will create efficiency-lowering costs of many pharmaceuticals and increase safety since human-based testing systems may better predict real-world outcomes.
Until last June’s Loper Bright decision, interpreting ambiguities, differences, and gaps in federal directives and legislation usually fell to federal agencies as enacted by the Administrative Procedure Act (APA), the 1946 federal law that governs how federal agencies create and implement rules and regulations and how courts review agency actions. This was governed by the Chevron deference, a two-step framework for courts to review how agencies interpret statutes in accordance with the APA.
The Chevron doctrine states that if a statute is clear, it should be applied as written. However, if statutes or legislation are determined to be ambiguous, courts should defer to an agency’s reasonable interpretation. The Loper Bright ruling overturned Chevron deference, allowing courts to exercise their independent judgment in deciding whether an agency has acted within its statutory authority. Courts need not defer to agencies’ interpretations of the law.
The Loper Bright decision is likely to significantly impact the life sciences industry by making it easier for companies to challenge federal agency regulations, potentially leading to increased litigation against the FDA. The ambiguity of FDA regulations has long been disputed. In 1989, the U.S. Court of Appeals for the District of Columbia was called to interpret the FDA’s requirement that a drug’s intended use have medical significance to be “effective in use.” The Court sided with the FDA, declining to examine “whether the statute compels the agency’s … reading” and “turn[ed] directly to the question whether the agency’s interpretation, as applied to this case, is permissible under the second step of Chevron.”
Post Loper Bright, courts are tasked with determining what “the statute compels” rather than making a simple determination of whether the FDA’s interpretations are “reasonable.” This could both present opportunities to challenge regulations and increase risks related to regulatory uncertainty and potential delays in product approvals.
In March, the first significant impacts of the Loper Bright decision on the life sciences industry were seen. The U.S. District Court for the Eastern District of Texas issued an opinion and judgment in American Clinical Laboratory Association v. FDA. The decision vacates and sets aside the Food & Drug Administration’s final rule, which was issued in May 2024, that would have required laboratories offering laboratory-developed tests (LDTs) to meet medical device requirements. The preamble to the LDT Ruling provided a multi-stage phase-out of the FDA’s enforcement discretion policy and ruled that the FDA lacks authority to regulate laboratory-developed tests. While the ruling was a victory for the American Clinical Laboratory Association, questions remain as to how the agency will proceed and the broader implications for regulation of lab tests and in vitro diagnostics in general.13
The implementation of tariffs aimed at supporting domestic industries is expected to create financial and operational challenges to downstream and ancillary companies. The life sciences industry is particularly sensitive to the effects of tariffs as increased pricing ripples through each stage of the manufacturing process, with disruptions manifesting in various forms, such as increased costs, supply chain uncertainties, and market competitiveness challenges.
The Food and Drug Administration shared data in 2019 that illustrated the reliance of the United States on foreign active pharmaceutical ingredient (API) manufacturers. The data revealed that 72% of API facilities supplying to the U.S. were overseas, with 13% in China. Additionally, 47% of all generic prescriptions in the United States are supplied by India.14 Imposing tariffs on pharmaceuticals has greater implications than just the economic impacts. Imposing tariffs may raise questions about compliance with World Trade Organization (WHO) rules. According to the 1994 Pharma Agreement, signed by Canada, the European Union, Japan, China, Norway, Switzerland, and the United States, most pharmaceutical products and substances used to produce them are exempt from tariffs.
The United States and the European Union in August 2025 released new details of their tariff agreement, which imposes a 15% tariff on pharmaceuticals. Prior to this agreement, the European Union faced the prospect of a 30% tariff. Pharmaceuticals account for roughly a quarter of U.S. imports from the EU as measured by total volume.15
47% of all generic prescriptions in the United States are supplied by India.
This abruptly changed as the U.S. administration announced in late September that a 100% tariff would go into effect for all U.S. pharmaceutical imports, entering the country effective October 1, 2025. The measure will not apply to companies building drug manufacturing plants within the United States.16
Geopolitics is also shaping the framework for tariff implementation. On July 31, 2025, the U.S. imposed a 25% levy on India in an executive order. A second order on August 6 doubled the initial tariff by adding 25% on imports from India, retributively for their purchase of Russian oil.17
The medical device industry will also be impacted by tariffs. In addition to imposing tariffs on medical devices that have historically been exempt from tariffs, the administration reinstated a 25% tariff on steel and certain steel derivatives and increased tariffs on aluminum from 10% to 25%. China is the biggest steel-producing country, accounting for 54% of world steel production in 2024.18 Both steel and aluminum are widely used in the medical field due to their unique properties. Stainless steel, particularly surgical steel, is known for its corrosion resistance and biocompatibility, making it ideal for surgical instruments and implants. The medical device industry’s heavily regulated nature creates challenges in finding alternative materials that meet the same safety and efficacy standards as these metals. Titanium is often used as a popular substitute for implantable devices. However, the cost of this alternative metal would still feel the impact of tariffs as China is the world’s largest producer of titanium.19
Nuclear verdicts, defined as an exceptionally high jury award in excess of $10 million that surpasses what should be a reasonable or rational amount, have increased exponentially over the past twenty-five years. Researchers speculate that not only can nuclear verdicts drive up the price of goods and services, but they may also adversely impact the availability of insurance and undermine the fairness and predictability of the rule of law.20
Nationwide, three case types made up two-thirds of nuclear verdicts in personal injury and wrongful death cases from 2013-2022: product liability (23.3%), auto accidents (23.2%), and medical liability (20.3%):
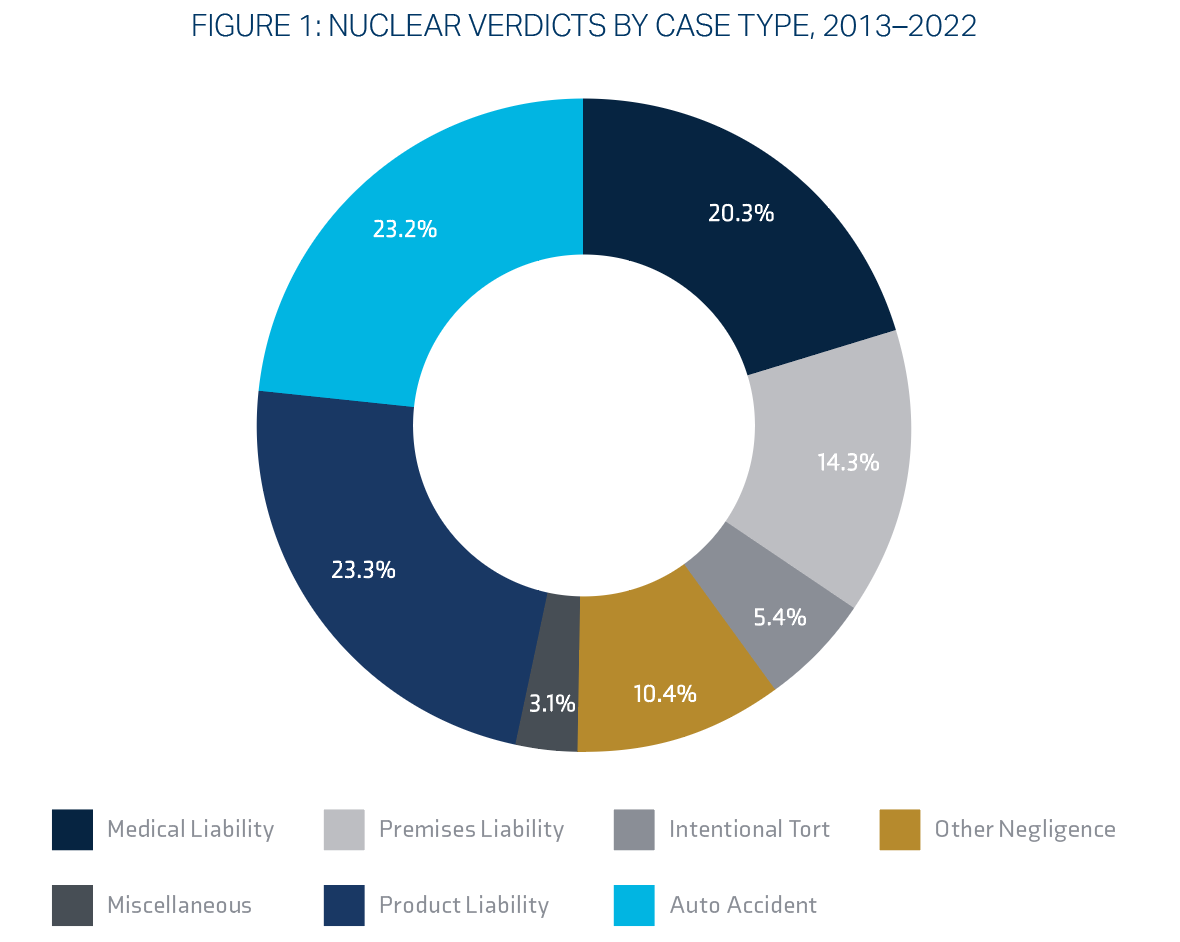
| Litigation Type | Mean | Median |
|---|---|---|
| Product Liability | $215.9 Million | $25.0 Million |
| Other Negligence | $99.8 Million | $20.0 Million |
| Intentional Tort | $94.6 Million | $28.6 Million |
| Auto Accident | $46.4 Million | $21.0 Million |
| Medical Liability | $33.6 Million | $19.6 Million |
| Premises Liability | $32.5 Million | $20.0 Million |
| Miscellaneous | $31.5 Million | $22.4 Million |
| All Personal Injury/Wrongful Death | $88.9 Million | $21.1 Million |
Source: Institute for Legal Reform: Nuclear Verdicts21
Between 2013 and 2022, the median nuclear verdict experienced a sharp rise, reaching $21 million, with some cases, particularly in product liability, resulting in even greater awards. In 2022, the median verdict in product liability cases alone peaked at an astounding $36 million — a 50% increase compared to 2013.22
Plaintiffs’ lawyers’ tactics are the primary drivers of large verdicts. Implementation of strategies like “reptile theory,” in which plaintiff’s attorneys seek to provoke an emotional response from jurors by framing the case in terms of public safety and danger, and jury anchoring, the suggestion of specific and often immoderate amounts for damages in the hopes of influencing jurors, are two tactics known to drive nuclear verdicts.23
Third‑Party Litigation Funding
Legal researchers have made a correlation between third-party litigation funding and nuclear verdicts. Third-party litigation funding allows hedge funds and financiers, including sovereign wealth funds and foreign interests, to invest in and control litigation within the United States, in exchange for a percentage of any settlement awarded. This practice is largely unregulated and is designed to maximize the profits for investors at the expense of the legal system, defendants, and consumers.
This practice has far-reaching implications beyond eroding jury awards for injured plaintiffs. Legal scholars argue that third-party litigation funding, in addition to disrupting and shifting the loyalty that counsel has from clients to investors, may pose a national security risk. The extent of foreign investment in U.S. litigations is unknown due to the industry’s lack of transparency, but the information available suggests that non-U.S. citizens, including sovereign wealth funds, participate in U.S. third-party litigation funding. This not only allows access to sensitive information but may also be a tactic to evade sanctions.24
A Bloomberg Law investigation found that A1, a subsidiary of Russian financial giant Alfa Group, financed lawsuits in New York and London, both before and after three of its founders were sanctioned following the 2022 invasion of Ukraine.25
New market entry, and what that could look like. Extremely competitive. Seven new national carriers entered the market. Higher limits due to overall capacity.
Commercial property insurance market trends for 2025 indicate a stabilizing market, offering more favorable conditions for many policyholders after years of significant rate increases. However, this shift is happening amidst persistent challenges, particularly for properties exposed to natural disasters and other high-risk factors. As a result of the Eaton wildfire in Altadena, California, in early 2025, insurers are increasing scrutiny and managing capacity more closely for properties with wildfire exposure and pushing for adequate insurance-to-value to ensure coverage limit accuracy.
Product liability trends in life sciences are marked by increasingly sophisticated litigation tactics from plaintiffs, such as using social media for targeted outreach and leveraging litigation funding. Regulatory shifts, like the new EU Product Liability Directive and AI Act, are introducing higher standards and potential exposures, while innovative technologies like AI in drug discovery and medical devices introduce novel liability questions. Companies face challenges in managing these complex risks, which are further influenced by factors like PFAS litigation and a tightening judicial scrutiny of scientific evidence in large-scale cases.
Despite these challenges, the product liability market has remained soft, driven by an increase in product liability capacity, with several well‑established carrier markets entering the life sciences segment in the last five years.
In light of recent and ongoing litigation relating to the release of ethylene oxide gas during the medical device sterilization process, and increased concerns of industrial runoff containing PFAS, there is increasing demand for life sciences companies to carry environmental and pollution liability coverage. With the exception of a pollution carve-back resulting from a hostile fire, a majority of commercial general liability and product liability policies fully exclude claims resulting from environmental pollution.
Where heightened environmental exposures exist, a robust prevention and response plan, in addition to a stand-alone pollution liability policy, remains the strongest risk mitigation strategy.
For publicly traded life sciences companies, whether newly public or with a longer-term track record, the risk of litigation is even more elevated compared to companies in other industries. In 2024, claims against life sciences companies accounted for 23% of all D&O claims, and in some years, this number has been closer to 25% or higher. Because of the historically high levels of litigation, there are fewer D&O carriers that will participate in the life sciences space, and those that do will oftentimes attempt to put forth punitive D&O program terms. This makes risk differentiation, strategic marketplace relationships, and the ability to secure competitive D&O program terms of utmost importance.
The regulatory landscape for medical device companies shifted significantly with the signing of the Consolidated Appropriations Act (H.R. 2617). The act mandated that the Food & Drug Administration institute express federal statutory cyber requirements for device manufacturers. The new statutes require device manufacturers to submit plans to the FDA outlining how device companies will identify, respond, and monitor post-market cybersecurity exploits and vulnerabilities.
A software bill of materials must be included for all off-the-shelf, open-source, and critical components that are part of the submitted device and commit to releasing post-market firmware, software, and patches throughout the device’s lifecycle.
Large healthcare data breaches continue to be reported to the Department of Health and Human Services’ Office for Civil Rights (OCR) in high numbers. As of January 28, 2025, the OCR data breach portal shows 725 data breaches of 500 or more records in 2024, the third consecutive year that more than 700 large data breaches have been reported to OCR.26
Insurance carriers remain vigilant in providing terms to risks that incorporate good cyber hygiene, such as multifactor authentication (MFA), vulnerability testing, and best-in-class cyber risk management.
The demand for increased regulatory fines and penalties coverage under life sciences cyber insurance programs has increased due to the new FDA cybersecurity statutes.

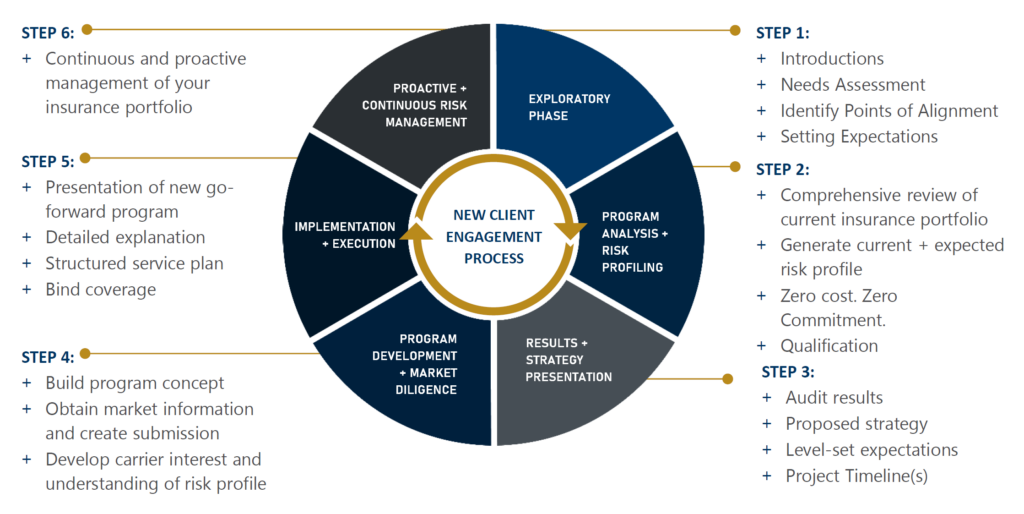
Partner with your broker early to prepare for any changes to increase renewal success.
It is important to collaborate with your broker’s industry experts who understand the business and the market for placing the specific risk. Collaborating with a team that can best represent your risk and partner with your operations is more important than ever during this disciplined market we are experiencing.
IMA has a team solely dedicated to managing cyber risks. They offer expert assistance, including coverage analysis, monetary loss exposure benchmarking, contract language review, in-depth cyber threat analysis, and strategic development of comprehensive, high-value cyber insurance programs.
Our contract review teams add value to our clients’ overall risk management program by ensuring the indemnity language is market standard and does not expose our clients to unforeseen losses that may not be insurable.
IMA invests heavily to deploy specialty niche teams concentrating on innovative technology, green energy initiatives, and advanced manufacturing. As every client is different, our Sustainability Advisory team provides clients with education, advice, and access to tools and best practices to advance their sustainability resilience and showcase their ESG performance for insurance underwriters.
Krista Hartin
VP, Life Sciences Segment Leader
Angela Thompson
Marketing Strategist, Market Intelligence & Insights
Brian Spinner
Sr. Marketing Coordinator, Market Intelligence & Insights
| Target | Industry | Aquiror | Value | |
|---|---|---|---|---|
| 1 | Intra-Cellular Therapies | Pharma | Johnson & Johnson | $14.7B |
| 2 | Inari Medical | Medical devices | Stryker | $4.8B |
| 3 | SpringWorks Therapeutics | Biotech | Merck KGaA | $3.7B |
| 4 | Endo | Pharma | Mallinckrodt | $3.4B |
| 5 | Mitsubishi Tanabe Pharma | Pharma | Bain Capital | $3.4B |
| 6 | Scorpion Therapeutics | Biotech | Eli Lilly | $2.5B |
| 7 | Nova Biomedical Corporation | Medical devices | Advanced Instruments | $2.2B |
| 8 | Efimosfermin alfa drug of Boston Pharmaceuticals | Pharma | GSK | $2.0B |
| 9 | Paragon 28 | Medical devices | Zimmer Biomet | $1.4B |
| 10 | IDRX | Pharma | GSK | $1.2B |
Source: PwC Analysis
*Data as of May 15, 2025. Reflects transactions announced through May 15, 2025 (some of which may not have closed yet). Note the Endo‑Mallinckrodt value was preliminarily calculated using the press release information as a value was not available from S&P Global Market Intelligence (and its affiliates, as applicable).